

Porcine Reproductive and Respiratory Syndrome (PRRS) is still, by far, the disease with the highest economic impact in the swine industry and its control is far from satisfactory. A better and more comprehensive picture of PRRSV variation and the monitoring of the circulation of new strains within a certain area/country/continent would certainly help veterinarians and producers to implement control and possibly eradication programs. To do so PRRSV sequencing became available worlwide starting from the late 1990’ mostly in North America, Europe and South-Eastern Asia.
The genome of PRRSV (see picture 1) is a single stranded RNA molecule which makes it prone to "make mistakes" (genetic mutation) during replication in the host. This "tendency to make mistakes" results is the presence in the field of different PRRSV strains, everyone unique in its own genetic sequence. Whether this sequence differences contribute to "different (clinical-pathological? immunological?) behaviours" is still a matter of debate among practitioners and researchers.

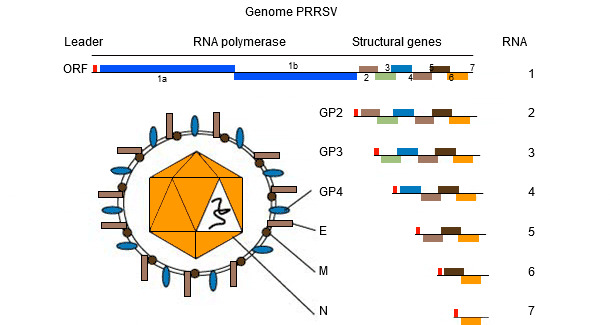
Basics of PRRS sequencing
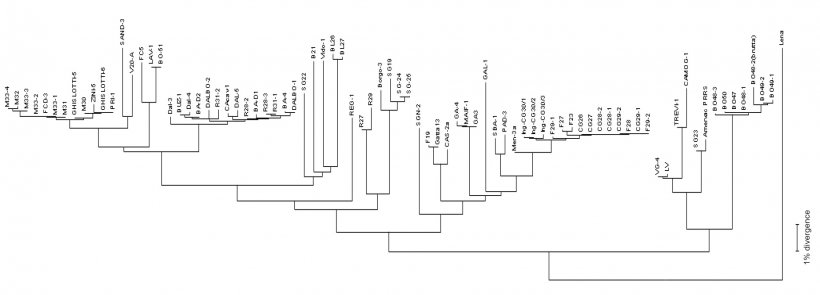
Virus sequencing is made on PCR products from field samples (sera, tissues, oral fluids) obtaining the reading of nucleotides usually from some viral RNA genome fragments (see picture 2) in target regions – ORFs (Open Reading Frame) and then comparing the homology percentage by phylogenetic analysis performed using dedicated softwares. The result of this process returns the degree of similarity (homology) between different PRRSV strains. Using graphical visualization softwares, also a dendrogram (or "phylogenetic tree") showing relatedness (or lack of) to reference virus sequence (see picture 3), can be obtained.

PRRSV genome encodes at least 10 ORFs. The most commonly used for sequencing – though they only represent 4% and 3% of the whole genome, respectively - are ORF5 (encoding the non glycosilated protein E) and ORF7 (encoding the nucleocapsid (N) protein). ORF5 represents a more variable region while ORF7 represents a more conserved region. Because of this, the same degree of variation (i.e. a 5% variation) found in ORF7 is more "dramatic" - in terms of genetic change – compared to ORF5 one. Interpretation of similarities (i.e. viruses are related or not) requires much more additional informations since the rate of genetic change can be highly variable.
It is extremely important to keep a record file of all sequences univocally identified and annotated to carefully match date, farm type (site 1-2-3), pig flow, location (GPS latitude/longitude) and origin of the sequence (type of animal/tissue/sample). To date, our PRRSV sequences dataset comprises more than 1300 ORF7 sequences from 2002. To interpret and make sense of differences it is even more important to match individual sequences to clinical events like the number of aborting sows and pre-weaning mortality in Sites 1 and mortality rate in Sites 2 and 3.
Practical Questions
Questions often asked by producers and veterinarians are:
- Do the observed genetic differences among the sequences represent normal variation of a single PRRSV strain in a farm/system, or represent multiple different strains present in a farm/system at the same time or in short temporal space of time?
- Is what I’m having now a "new outbreak" caused by a "new strain" or is it a re-circulation?
To answer these questions we must agree on the accepted degree of homology of two viral strains collected within a certain period of time (12-24 months?). In other words the similarity cut-off. A 97-98% of sequence homology or 2-3% difference is a generally accepted value. According to my experience is rather difficult to see a change higher than 2% in a "clinically stable closed population" (a conventional sow herd or a pig flow) as we observed obtaining the "same strain" for a period as long as 3 years in a single clinically stable herd. Opposite to this, every time we noticed a consistent PRRS activity a "new" and phylogenetically diverse (90% homology or less) strain is recovered. Unfortunately we do not absolutely know if these big differences, that we sometimes observe, are the result of a sudden virus change/mutation (unlikely in my personal opinion) or the introduction of a new strain. What is clear and well accepted is the fact that genetic similarity/diversity is in no way predictive of immunological similarity (i.e. indicative of cross protective immunity) and is not at all predictive of intrinsic pathogenicity (does not tell if a particular strain is "good" or "bad").
Whole genome sequences that are today available (unfortunately still more for research purposes than for everyday diagnostic usage) will certainly help to answer this question.
It is very important to analyze new PRRSV sequences against a broad reference set representing the farm, the system and the region, as well as the sequences of available commercial vaccines (this will make it possible to differentiate between field and vaccine strains). At the moment we are still using a set of open-source softwares managed by Padova University to build our phylogenetic trees and keep them organized by pig flow within our total production system; we could also be joining soon two other "ad hoc computer programs" (Bioportal by University of Davis-California and CLASSIFARM-PATH by IZSLER (Brescia, Italy) that will have a much larger set of sequences to compare with allowing to better understand PRRSV circulation in Italy and possibly in EU.
Acknowledgement: Thanks to Prof. Michele Drigo (UNI-PD) for the interesting discussion and revision of this paper.



")
