

The pathogenesis and the lesions caused by PRRSV infection is largely dependent on the common characteristics of the viruses belonging to the Arterivirus genus: (i) capability to enter and replicate in their primary target cell, the macrophage; (ii) prolonged viraemia after infection; (iii) long lasting, persisting (not persistent!) infection within the animal. The acute phase of viraemia in growing pigs lasts for 4 weeks approximately, after that the virus will be found predominantly in lymphoid tissues – usually for the rest of the life of a production animal – and viraemia can re-occur periodically (reviewed in details in Chand et al. 2012, Curr Opin Virol).
In the lung tissue, lesions induced by respiratory viruses can develop by two main routes: direct destruction, dysregulation of the pneumocytes, and/or infection of inflammatory and immune cells that induce tissue damage by the release of active cytokines, noxious metabolites. These activated cells can recruit other inflammatory cells to the site of infection that may augment tissue damage, but they have an important role in viral clearance, and regulation of the inflammation as well.

In case of PRRSV infection the primary target cell is the alveolar macrophage. The tissue damage is the consequence of direct apoptosis (and necrosis) of these cells and – to a greater extent – their neighboring (bystander) cells due to the release of apoptotigenic cytokines, reactive oxygen species, and nitric oxide. At the same time proinflammatory cytokines are also released, that are responsible for the recruitment of other inflammatory cells, and some of them are causing the systemic symptoms (fever, lethargy etc.). Anti-inflammatory, regulatory cytokines are also secreted. It is also well documented that similarly to other viruses PRRSV has the ability to suppress the primary type I interferone response – a strong antiviral mechanism of the innate immunity – in early stages of infection through their non structural proteins giving the potential to replicate and spread more efficiently. Different PRRSV isolates however have different immunemodulatory capabilities, and the clinical signs, the lesions and the overall outcome of the infection is largely dependent on the shifted balance of the aforementioned mechanisms. The damage/destruction of the macrophages can give rise to secondary infections as it is commonly observed in field conditions.
In case of experimental infections conducted in isolated facilities the clinical signs and the lesions observed are largely dependent on the pathogenicity of the strain used for the inoculation of the animals. The most severe respiratory lesions can be observed 7–14 days PI. Macroscopically they are consolidated, tan, mottled areas, affecting more severely the cranioventral lobes, but they can be found scattered in the entire lung tissue (Picture 1). The most relevant histopathological lesions include: (1) pneumocyte hypertrophy and hyperplasia, (2) septal mononuclear infiltration, (3) intraalveolar necrotic debris, (4) intraalveolar inflammatory cell accumulation and (5) perivascular inflammatory cell accumulation.
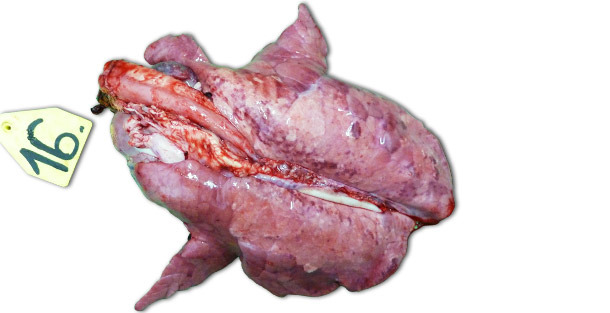
Picture 1. Lungs of a piglet exterminated 14 days PI, inoculated with higly pathogenic Type I subtype 3 strain “Lena”.
In a challenge study where pigs were euthanized 14 and 21 days PI, significant decrease of intraalveolar necrotic debris and intraalveolar inflammatory cells was observed by 21 days PI in the infected pigs; in contrast, only minimal change was present for the other three categories. These findings can be explained by the fact that formation of intraalveolar necrotic debris and subsequent accumulation of intraalveolar inflammatory cells (predominantly neutrophil granulocytes) reflects the acute phase of the disease (Picture 2), where extensive lung tissue damage is caused by the release of harmful substances from virus infected macrophages. After the initial, acute stage of the disease – in the absence of secondary bacterial infection – the immune system and natural healing mechanisms remove the necrotic tissue, and cells of the acute inflammation (neutrophil granulocytes) disappear, alveoli are freed of content and damaged pneumocytes are replaced by proliferating type II pneumocytes.
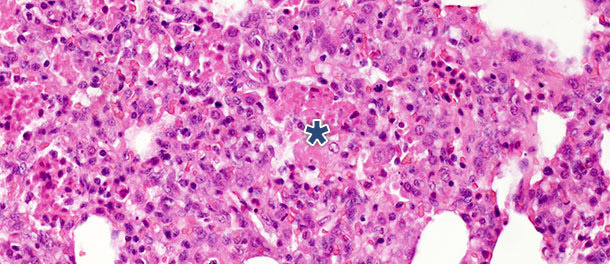
Picture 2. Intraalveolar necrotic debris, and inflammatory cell accumulation (asterisk) in the lung tissue
at 10 days PI with PRRSV Type 1 subtype 1 isolate. H.E. 200×
Type II pneumocytes are cuboidal cells typically located at the insertion of the alveolar septa. In mammalian lung tissues they constitute 60% of all alveolar epithelial cells, but cover only about 5% of the alveolar surface. Their most important role is the synthesis, secretion and recycling of pulmonary surfactant which reduces alveolar surface tension, preventing from collapse during expiration. The other important function of the type II pneumocytes lies in their proliferative potential. After injury of type I pneumocytes, type II pneumocytes serve as progenitor cells to replace and eventually differentiate into previously damaged and desquamated type I pneumocytes. In the challenge study described above, the number of these cells increased significantly by 10 days PI (Picture 3) and did not decrease by 21 days PI. Though the study was terminated at 21 days PI and total histological healing (restitutio ad integrum) was not observed in the infected group, the animals recovered from the clinical disease and the elevated number of type II pneumocytes after inoculation in both early and late stages of the disease demonstrates their importance in lung tissue healing (discussed in details in Balka et al. 2013. J Comp Pathol).
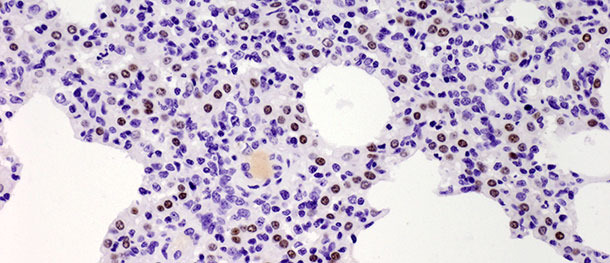
Picture 3. Brown dots indicate the nuclei of the Type II pneumocytes identified with anti-TTF-1 antibodies. Significant increase in the number of positive cells was observed at 10 days after infection with a Type I subtype 1 isolate. IHC 200×



")
