

Viral DNA acts as a fingerprint, and comparing the outbreak sequence with international reference genomes is done to establish genetic relationships between outbreaks.
To classify it, the harmonized molecular characterization scheme developed by the EU Reference Laboratory for African swine fever (EURL-ASF, CISA-INIA/CSIC) was applied, combining a multigene approach with whole-genome sequencing.

Characteristics of the ASF virus
The ASF virus:
- Has double-stranded DNA
- Is exceptionally large and complex, with approximately:
- 170,000 to 194,000 nucleotides
- ± 170 genes, depending on the viral isolate
- Exhibits high genetic conservation, especially in the central region of the genome
How are ASF viruses classified?
Level 1. Global genotypic classification of the ASF virus
The ASF virus has traditionally been classified using the sequence of a gene called B646L, which encodes the structural protein p72. Based on differences in this gene, we can establish a first level for classifying the viruses, which has allowed us to identify 24 genotypes worldwide.
Despite this global diversity, since its introduction to Georgia in 2007 from East Africa, genotype II has been the main driver of the current ASF pandemic worldwide, affecting numerous countries in Europe, Asia, and Oceania, and reaching the Dominican Republic and Haiti in the Americas in 2022.
Currently, genotype II is the only one circulating in Europe, both in wild boar and domestic pig populations.
Viruses within genotype II are extremely similar genetically, so this first level of classification is insufficient to discriminate between closely related viruses and to investigate the origin and relationship between outbreaks on a regional or local scale.
Level 2. Multigene approach to intra-genotypic differentiation of ASF viruses
To overcome the limitation posed by the high genetic similarity of the different viruses belonging to genotype II, a multigene approach was developed based on sequencing 6 additional regions of the viral genome.
- The intergenic region (IGR) between genes I73R and I329L
- The central variable region (CVR) of gene B602L
- Gene O174L
- Gene K145R
- The intergenic region between genes 9R/10R of the 505 multigene family (MGF)
- The region designated ECO2, located between genes I329L and I215L
This approach allows for the joint analysis of genetic variants present in different regions of the genome, including short tandem repeats (SRTs) and point mutations (SNPs – single nucleotide polymorphisms), providing a significantly higher resolution than that obtained using individual markers.
The specific combination of variants detected in each of these regions defines a characteristic genetic signature for each virus.
Based on this, up to 28 genetic subgroups have been defined within the genotype II currently circulating in Europe. Genetic subgroup 1 corresponds to the basal profile of genotype II and is associated with the reference strain VPPA Georgia 2007/1.
This classification has a strictly epidemiological purpose, oriented towards molecular traceability and the analysis of virus dispersal routes. It does not imply known differences in virulence, transmissibility or clinical presentation.
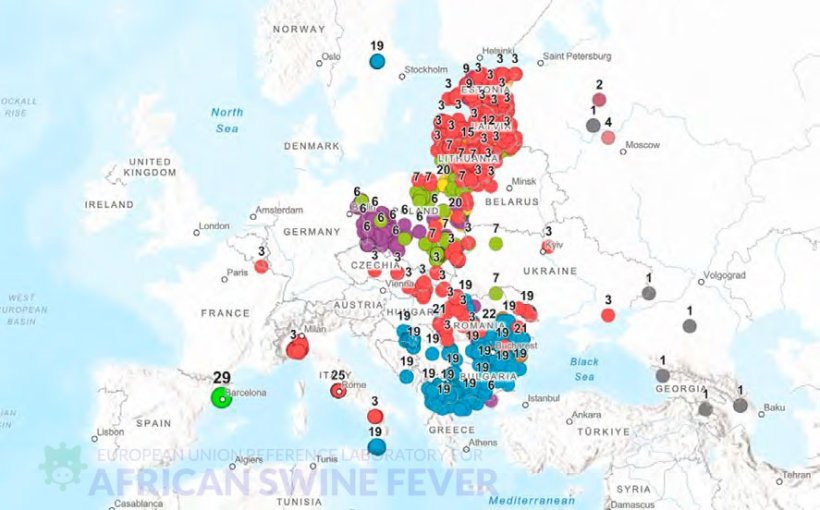
The virus detected in wild boar in the Collserola area, near Barcelona, showed genetic differences compared to previously described viruses. Due to these differences, it has been classified within a new genetic group: group 29 within genotype II (Table 1).
Table 1. Genetic subgroups of ASF virus genotype II defined by the multigenic approach: genetic profile.
| Subgroup | CVR | IGR I73R / I329L | O174L | K145R | IGR MGF 505 9R/10R | ECO2 |
|---|---|---|---|---|---|---|
| 1 | I | I | I | I | I | I |
| 2 | I | I | I | I | II | I |
| 3 | I | II | I | I | I | I |
| 4 | I | I | I | I | III | I |
| 5 | II | II | I | I | I | I |
| 6 | I | II | II | II | I | I |
| 7 | I | II | I | II | I | I |
| 8 | I | II | I | II | II | I |
| 9 | I-SNP1 | II | I | I | I | I |
| 10 | I | I | II | II | I | I |
| 11 | I-SNP1 | II | I | II | I | I |
| 12 | I | II | I | I | II | I |
| 13 | I | III | II | II | I | I |
| 14 | I-SNP1 | II | I | I | I | I |
| 15 | I | II | I | I | V | I |
| 16 | I | II | I | I | IV | I |
| 17 | I | II | I | I | I-V1 | I |
| 18 | I | III | II | I | I | I |
| 19 | I | II | I | I | I | II |
| 20 | I | IV | I | II | I | I |
| 21 | I | II | II | I | I | I |
| 22 | I | II | II | I | I | II |
| 23 | I | II | I | I | VII | I |
| 24 | I | II | I | I | VI | I |
| 25 | I | II | I | II | VIII | I |
| 26 | I | II | I-SNP1 | I | I | I |
| 27 | I | II | I | II | V | I |
| 28 | I | II | I | I | IX | I |
| 29 | I | II | I | I | I-V2 | I |
The Spanish isolate has a genetic profile closely related to the basal profile of genotype II (Georgia 2007), coinciding in five of the six regions analyzed and differing in one. In the intergenic region MGF505-9R/10R, a mutation (G→A) was detected that was not previously described among the 1,186 genotype II viruses available in the EURL-PPA database or in international databases.
What do the variants mean?Within each region analyzed, the variants are named in a standardized way:
In this way, we can differentiate similar viruses with a high level of detail, maintaining consistency between studies. |
Evaluation of the genetic coverage of ASF virus genotype II in Europe
Comparison with other outbreaks requires access to up-to-date and representative genetic information on the viruses circulating in Europe.
Countries were categorized based on the genetic coverage available in the European Union Reference Laboratory for ASF sequence database and official outbreak notification data from domestic pigs and wild boar.
| Category | Year of the last genetically characterized isolation | Total number of sequenced viruses available | Existence of active circulation of the ASF virus | Official declaration of eradication where applicable | Countries |
|---|---|---|---|---|---|
| 1 | Until 2025 | Representative number | Active circulation | Estonia, Greece, Croatia, Italy, Romania, Moldova | |
| 2 | Recent genetic information (2023–2024) | Sequences associated with the latest epidemiological episode are available | Active or countries where ASF has been eradicated | Belgium, Bulgaria, Czech Republic, Germany, Latvia, Slovakia, Sweden, Albania, Bosnia and Herzegovina, North Macedonia, Montenegro, Kosovo | |
| 3 | Limited genetic data (2021–2022) | Active circulation | Lithuania, Poland | ||
| 4 | Lack of genetic data in the last five years or a very small number of isolates available | Active circulation | Hungary, Serbia, Ukraine, Russia, Armenia, Azerbaijan, Belarus, Georgia | ||
This classification highlights the existence of relevant geographical and temporal gaps in the genetic information available in Europe.
Table 3. Number of African swine fever virus genotype II isolates and corresponding percentage, calculated on a total of 1,187 sequences for which complete information of the six genomic regions is available.
| Genetic group | Geographic distribution (year) | Nº | % |
|---|---|---|---|
| 1 | Georgia (2007), Armenia (2007–2008), Azerbaijan (2008), Russian Federation (2009, 2012, 2019), Poland (2022) | 11 | 0.9 |
| 2 | Russia (2012) | 1 | 0.1 |
| 3 | Ukraine (2012, 2019), Belarus (2013), Lithuania (2014–2018, 2020–2022), Poland (2014, 2018, 2021–2022), Latvia (2014–2015, 2017–2024), Estonia (2014–2025), Czech Republic (2017–2018), Romania (2017–2019, 2021, 2023–2024), Moldova (2017–2018, 2025), Hungary (2018–2019), Belgium (2018), Slovakia (2019–2020, 2022–2023), Italy (2022–2023), Russia (2019) | 439 | 37.0 |
| 4 | Russia (2012) | 1 | 0.1 |
| 5 | Estonia (2015–2016) | 7 | 0.6 |
| 6 | Poland (2016, 2019, 2021–2022), Germany (2020–2024), Czech Republic (2022, 2024) | 154 | 13.0 |
| 7 | Poland (2016–2019, 2021), Lithuania (2017–2018, 2020–2022), Romania (2019), Latvia (2021, 2023–2024), Russia (2018) | 156 | 13.1 |
| 8 | Poland (2016–2017) | 11 | 0.9 |
| 9 | Estonia (2017) | 10 | 0.8 |
| 10 | Poland (2017) | 2 | 0.2 |
| 11 | Poland (2017) | 1 | 0.1 |
| 12 | Lativa (2017–2018, 2021–2024) | 29 | 2.4 |
| 13 | Poland (2017) | 1 | 0.1 |
| 14 | Lithuania (2017), Latvia (2023–2024) | 4 | 0.3 |
| 15 | Lithuania (2017) | 1 | 0.1 |
| 16 | Lithuania (2017–2018) | 5 | 0.4 |
| 17 | Latvia (2017–2018) | 3 | 0.3 |
| 18 | Poland (2018) | 1 | 0.1 |
| 19 | Romania (2018, 2021, 2023–2025), Bulgaria (2018–2020, 2024), Serbia (2019–2020, 2022), Greece (2020, 2024–2025), North Macedonia (2022, 2024), Italy (2022–2023), Sweden (2023), Croatia (2023–2025), Bosnia and Herzegovina (2023), Montenegro (2024), Albania (2024), Kosovo (2024), Moldova (2025) | 268 | 22.6 |
| 20 | Poland (2018–2019, 2021–2022) | 23 | 1.9 |
| 21 | Romania (2019), Czech Republic (2024) | 8 | 0.7 |
| 22 | Romania (2019) | 12 | 1.0 |
| 23 | Latvia (2020) | 1 | 0.1 |
| 24 | Romania (2021) | 1 | 0.1 |
| 25 | Italy (Lazio, 2023) | 12 | 1.0 |
| 26 | Italy (Piedmont, 2023) | 4 | 0.3 |
| 27 | Poland (2021–2022) | 15 | 1.3 |
| 28 | Estonia (2022–2024) | 5 | 0.4 |
| 29 | Spain (2025) | 1 | 0.1 |
Level 3. Complete sequencing of the genome of the virus detected in Spain
The complete genome of the Spanish isolate was sequenced with the aim of obtaining the highest level of genetic resolution.
The proposed nomenclature for the strain found in Barcelona is SP25WB2611 and the sequence is available from GenBank at this link.
The analysis revealed that the isolate has:
- a total length of 180,757 nucleotides
- an approximate reduction of 9.8 kb due to a large deletion. This deletion involves the complete loss of 21 genes, notably genes from multigene families (MGFs). In particular, several genes from the following families:
- MGF110 (MGF110-7L, -8L, 9L, -10L/14L, -12L, -13La y -13Lb)
- MGF360 (MGF360-4L y MGF360-6L)
- GF100 (MGF100-1R)
Deletions in this region, although not identical in extent or genetic content, have been described in both Europe and Africa, indicating that this type of structural reorganization is part of the natural evolutionary dynamics of the virus.
Outside the deleted region, the Spanish isolate shows high genetic stability compared to the reference strain genotype II Georgia 2007/1. Comparative analysis revealed:
- Nucleotide identity greater than 99.9% was found in all regions shared between the two genomes.
- Compared to Georgia 2007/1, the following were identified:
- 18 single point mutations (SNPs).
- 13 short insertions and deletions (INDELs) of less than 5 nucleotides.
- A 5-nucleotide deletion located in a gene of the MGF505 family is noteworthy; this deletion was not identified in any of the isolates available in public databases.
Scenarios about the origin of the virus
To date, it has not been possible to directly establish a specific geographic or epidemiological origin for the outbreak. The presence of a unique genetic signature in the Spanish isolate allows us to rule out its relationship with genotype II sequences currently available in public databases.
Accidental release from a research laboratory
To assess the hypothesis of a possible accidental release from a research laboratory, 81 samples of viruses used in experimental activities at CReSA-IRTA were analyzed at the national reference laboratory. None of them presented the specific genetic markers (SNPs) identified in the Spanish virus that caused the outbreak.
The results obtained showed no genetic match between the Spanish isolate and the viruses used in experimental activities at CReSA-IRTA, neither at the level of partial markers nor at the whole genome scale.
Introduction from active European foci through natural or progressive transmission
This is not considered likely, as the nearest active European outbreak at the time of detection (Italy) was located more than 500–600 km away, and the countries between them have robust ASF monitoring programs. Furthermore, the genetic profile of the virus detected in Spain did not show a close relationship with the Italian viruses.
Deliberate introduction
In a hypothetical scenario of deliberate introduction, it is inconsistent to use a strain like the Spanish isolate, which has a high deletion rate and uncertain biological behavior, reducing its predictability in terms of transmission and pathogenicity. Deliberate introductions are usually associated with well-characterized strains with known epidemiological behavior.
Long-distance introduction mediated by human activities
This route is the most common mechanism for the spread of ASF virus over long distances and is widely documented in the epidemiology of the disease. This scenario is consistent with several epidemiological elements observed in the outbreak detected in Catalonia:
- Isolated outbreak.
- Absence of intermediate outbreaks in neighboring countries.
- Outbreak located in a highly connected environment with high human movement and a dense road and rail infrastructure network.
- Genetic divergence from dominant lineages in Europe, including the closest relatives, suggesting an introduction unrelated to known geographic expansion.
The affected area adds elements that increase the plausibility of introduction via waste. The initial outbreak was located in an environment where urban centers are embedded in forest and agricultural land, there is a stable presence of wild boar populations without significant physical barriers, and they have access to household waste containers, picnic areas, busy parks, peri-urban areas, and road and rail infrastructure.
Report prepared by the SCIENTIFIC COMMITTEE FOR ADVISORY SUPPORT ON THE OUTBREAK OF AFRICAN SWINE FEVER IN SPAIN.
Download the pdf of the report (in Spanish) 
Maria del Carmen Gallardo Frontaura
Marta Martínez Avilés
Christian Gortázar Schmidt
Carlos Sánchez García-Abad
Antonio Palomo Yagüe
Daniel Babot Gaspa
Jesús Salas Calvo
Emilio García Muro
Ana Rodríguez Castaño
This preliminary report covers matters known up to 31.01.2026
