

Influenza A virus (IAV) is one of the most important respiratory pathogens of swine. It impacts mortality and causes significant financial losses through decreased production and the costs associated with vaccination and treatment. Further, due to the susceptibility of swine to transient infection with IAV from different species, namely humans, novel reassorted viruses may emerge that have implications for public health. Consequently, understanding the genetic diversity of circulating viruses in swine can identify novel viral lineages, and provide criteria for informing and improving intervention strategies.
The genetic diversity of swine IAV in the United States represents antigenic drift and shift along with introduction of IAV from other species into swine populations (Vincent et al., 2008). Broadly, there is the co-circulation of three dominant subtypes (H1N1, H1N2, and H3N2) and four major genetic lineages described by the origin of the gene segments. The classical H1 swine lineage originated from the 1918 human Spanish flu pandemic. The second major lineage was detected in the late 1990s with the hemagglutinin (HA) and neuraminidase (NA) derived from human seasonal H3N2 influenza. This lineage was a novel reassorted virus, containing HA, NA, and PB1 gene segments derived from seasonal human H3N2 influenza, PB2 and PA gene segments derived from avian influenza, and NP, M, and NS gene segments from classical H1N1 swine influenza, thus called “triple reassortant.” These viruses reassorted further with classical H1N1 viruses to acquire the HA and/or NA, resulting in new genetic types of H1N1 and H1N2 viruses. The “triple-reassortant internal gene” (TRIG) constellation remained relatively consistent with swine (M, NP, and NS genes), avian (PB2 and PA genes), and human (PB1) influenza virus origins. The third lineage resulted from repeated spillovers of human seasonal H1 IAV viruses into swine herds early in the twenty-first century, creating two distinct lineages of human seasonal H1N1 and H1N2 viruses. The fourth and newest lineage represents a modern human seasonal H3N2 spillover that occurred in 2010-11. Within these four major HA lineages, the viruses continue to evolve, forming new genetic clades (Figure 1).

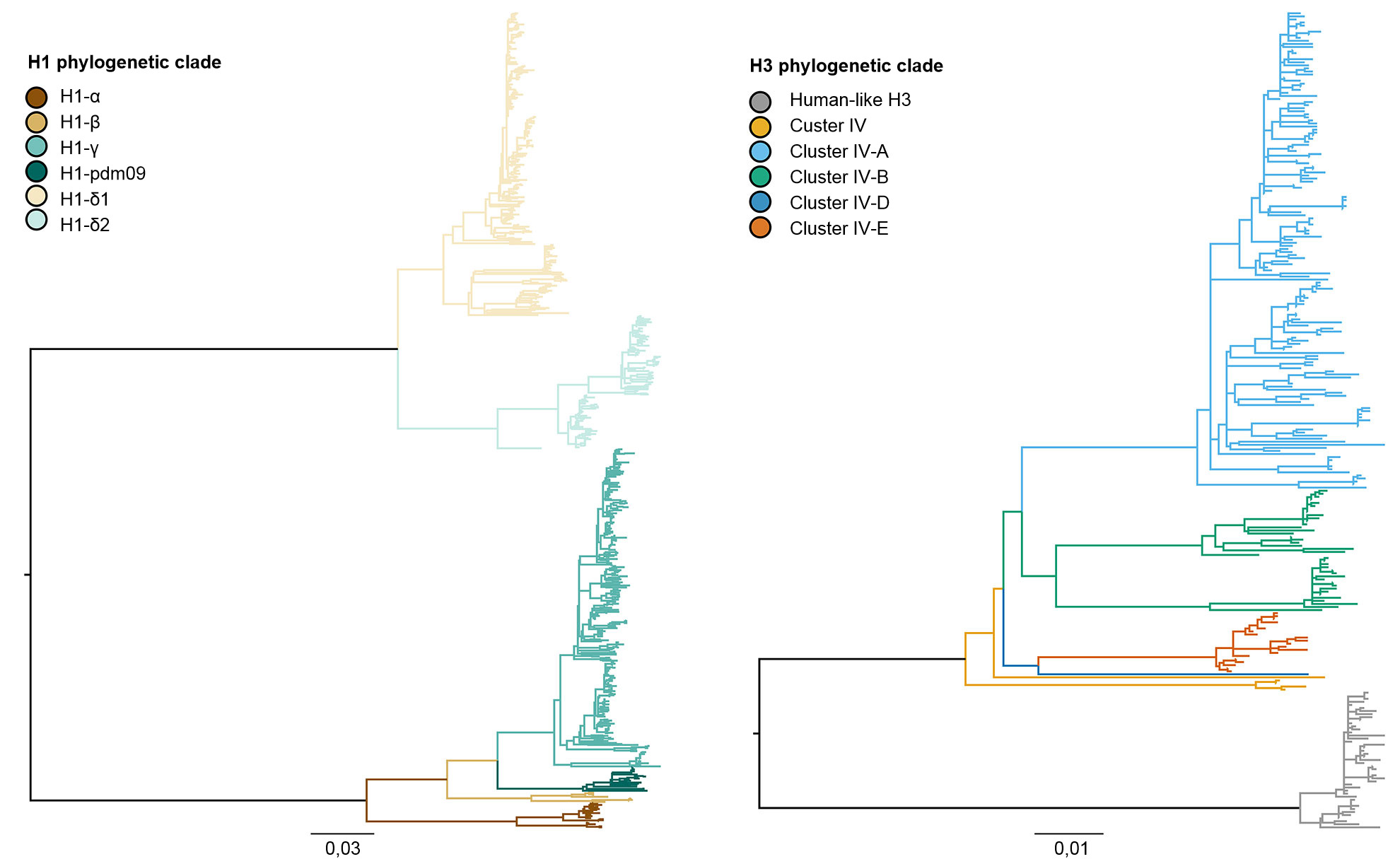
Figure 1. Phylogenies describing the genetic relationships of swine H1 and H3 influenza A hemagglutinin gene sequences from 2015 generated using maximum likelihood methods. Branch color represents clade designations. All branch lengths are drawn to scale, and the scale bar indicates the number of nucleotide substitutions per site.
In a capacity-building effort to understand the genetic diversity of swine IAV and monitor the 2009 H1N1 pandemic (H1N1pdm09) virus in swine, a national surveillance system was initiated in 2009 by the United States Department of Agriculture (USDA) and implemented through the National Animal Health Laboratory Network (NAHLN). The system relies upon anonymous submissions from producers and veterinarians from routine diagnostic investigations; however, limited fine-scale spatial information regarding the provenance of the sample is shared. Epidemiological data collected includes the US state of origin, sample type, reason for submission, age class, location type, test results and, if applicable, the HA, NA, and M genes are sequenced and deposited into the National Center for Biotechnology Information’s online sequence repository, GenBank (Korslund et al., 2012; Anderson et al., 2013). These data, and the continued investment in the system, has provided insight into the patterns of swine IAV spread, genetic diversity throughout the year, and the dynamics of IAV evolution in North America from 2010 to present (Anderson et al., 2013; Anderson et al., 2015; Lewis et al., 2014; Rajão et al., 2015).
The three influenza virus subtypes (H1N1, H1N2, and H3N2) endemic in the U.S. swine population have been detected every year from 2010 to present. The H1N1 and H1N2 subtypes were detected at similar frequencies (~35%) across the 5 years. The H3N2 subtype represented ~30% of the identified viruses during the total time period. To describe the genetic diversity of the H1 viruses, a system of greek nomenclature has been used to divide the clades of HA data into classical lineage (α, β, γ, γ-2, and H1N1pdm09) or the human lineage (δ-1, δ-2). Similarly, the H3 cluster IV genes separated into 6 genetic clades (A through F), and a human-like H3 clade (Rajão et al., 2015; Kitikoon et al., 2013). However, despite the potential for 14 genetic clades to circulate, much of the observed HA diversity in US swine herds was restricted to three clades. From January 2015 to December 2015, 43% of H1 isolates were from the γ-clade in the classical lineage and 37% were classified as δ-1 from the human-seasonal lineage; the remaining 20% of H1 viruses came from the δ-2, α, H1N1pdm09 and β clades. Of the 8 potential H3 clades, cluster IV-A represented the majority of circulating swine H3 IAV in production herds (61% of H3 viruses during 2015), a growing number of isolates were identified as human-like H3 (from 5% in December 2014 to 25% in December 2015), with the remaining isolates representing sporadic detections of clusters IV-B, IV-C, IV-D, IV-E.
In the context of effective vaccination programs, there is a renewed awareness that the neuraminadase (NA) may also have an important role due to its increasing diversity (Sandbulte et al., 2016). Though this complicates the situation, the NA has far fewer lineages compared to the HA genetic lineages. The HA are paired with N2 genes derived from one of two human H3N2 seasonal-lineages (either from 1998 or 2002: (Nelson et al., 2011)), N1 genes from the classical swine-lineage, or human H1N1 seasonal-lineage N1 genes (Anderson et al., 2013). In the currently circulating swine IAV, the N1 is generally of classical-lineage (94%), and the N2 is generally a 2002-lineage (83%), with the 1998-lineage N2 consistently detected, but representing a smaller component in IAV isolates.
A second concern for intervention efforts is associated with spatial patterns in the diversity of swine IAV. The US is divided into five different regions for reporting purposes based on USDA-APHIS veterinary services districts (Figure 2A). There are subtle and important differences between regions in diversity and in raw abundance of samples provided to the USDA surveillance system (Figure 2B). While Regions 1 and 2 both have higher detections of H1N1, Region 3 has more H1N2 and Region 4 has more H3N2. Despite a swine population of 1.6 million pigs (USDA-NASS 2012), very little data has been collected through the US surveillance system for Region 5. Region 2 shows the most diversity in terms of the different HA and NA clades observed, and most of the swine IAV isolates are submitted from this region. Overall, the HA and NA clade distributions suggest that regional or local decisions on vaccine components may be of value.
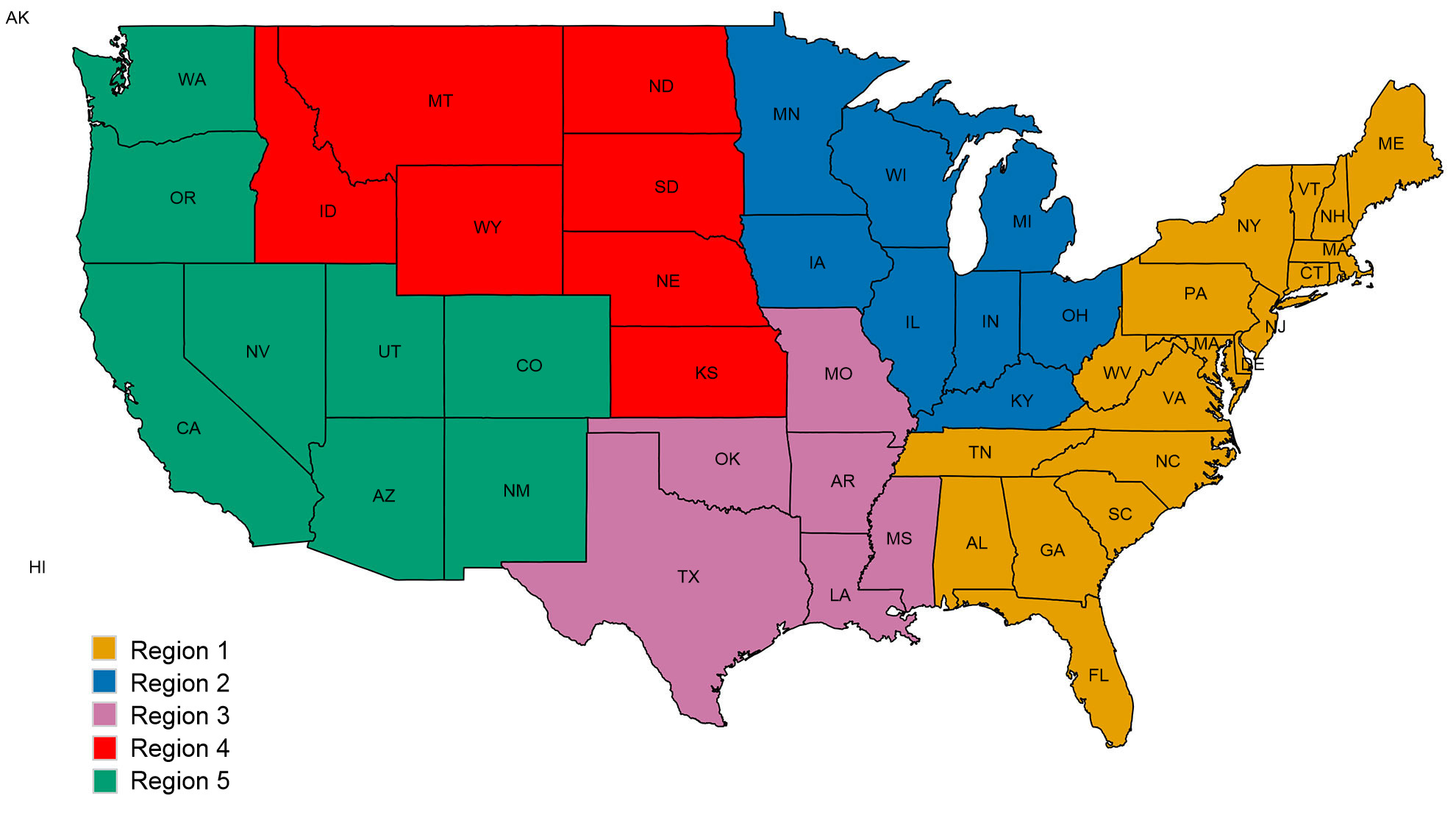
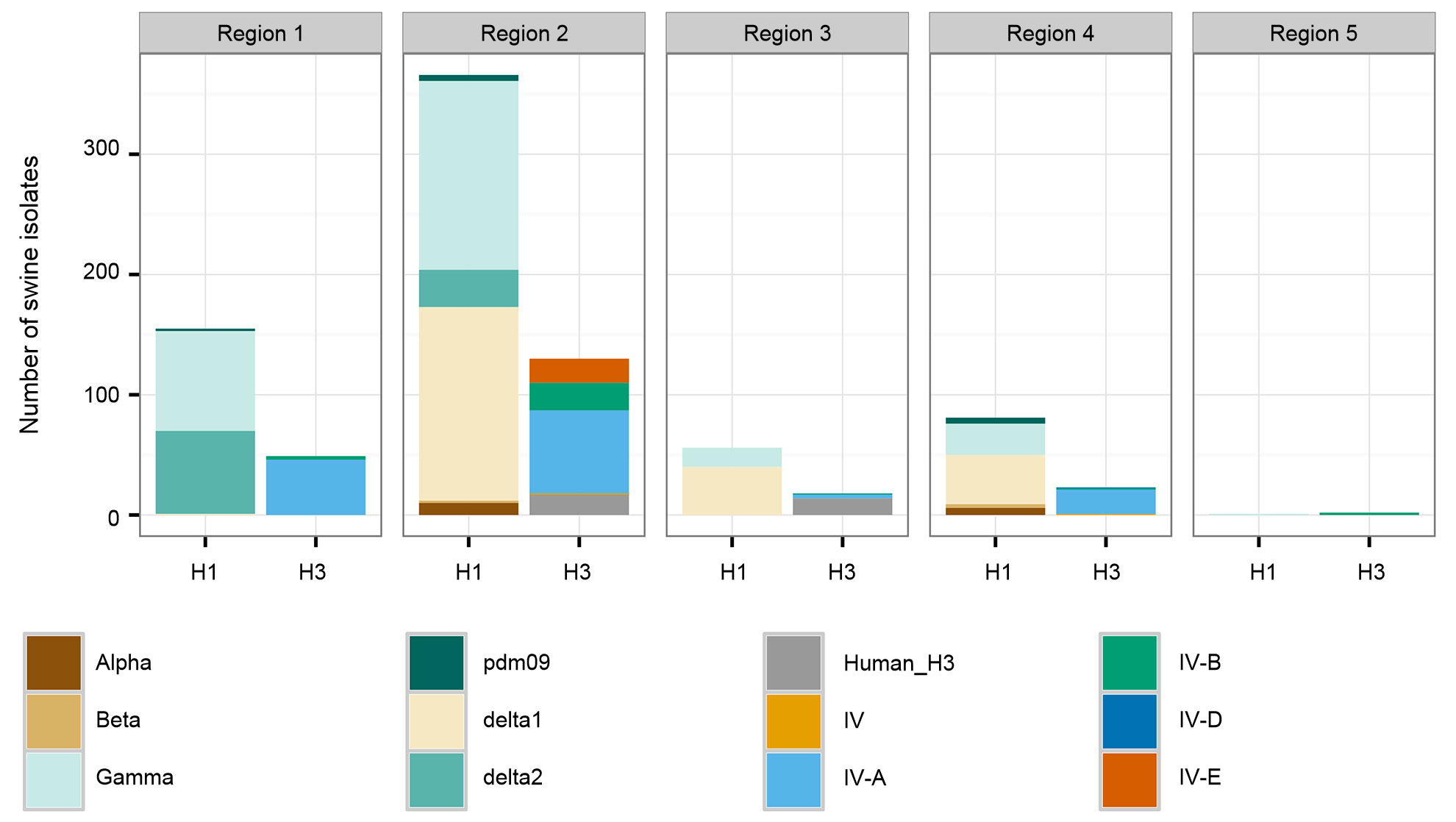
Figure 2. United States Department of Agriculture, Animal and Plant Health Inspection Service (USDA-APHIS) veterinary services regions (A), and the number of swine influenza A isolates, classified by phylogenetic clade and colored as in Figure 1, collected from each region during 2015 (B).
The genetic diversity of swine IAV is a complex issue at regional and especially at global levels. In the United States, there are 17 genetic clades that have emerged and persisted following spillover events from non-swine (namely human) hosts and subsequent ecological and evolutionary processes. Though consisting of different genetic clades derived from different non-swine spillover episodes, a similar level of genetic diversity exists in global swine populations (e.g., Watson et al.,2015; Bahl et al.,2015; Vijaykrishna et al. 2015). The genetics of contemporary swine IAV can be used to inform studies on antigenic evolution and diversity, and these analyses should be conducted with contemporary data sourced from regional populations. Together these data, and with the implementation of appropriate vaccination platforms, provide fundamental data for use in the selection of vaccine strains and informs risk management policies for agricultural and public health.
