

Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) has a single stranded RNA genome which makes it highly prone to genetic mutation. This also makes every PRRSV strain unique, so that genetic typing is a useful method for disease diagnosis and control. Diagnostic genetic typing is made by determining the order of nucleotides within a DNA copy of the viral RNA genome fragment – DNA sequencing. Today the most often used fragment for this purpose is ORF5, the gene encoding the major envelope glycoprotein, mainly because it shows extensive genetic diversity.

Diagnostic DNA sequencing
Discrimination between PRRSV Type 1 (European) and Type 2 (American) is easy to accomplish by most diagnostic PCR assays. Discrimination between individual strains from each of the two genotypes requires DNA sequencing. To do this, body fluid or tissue with moderate to high amounts of PRRSV are treated to isolate RNA, that is then copied to DNA by reverse transcription, and the ORF5 gene is amplified by PCR, and submitted to a DNA sequencing facility. This process is mostly automatic and generally takes one to three days. The raw sequence data files are sent to the diagnostic laboratory for analysis. The report typically includes the nucleotide sequence of the strain, its similarity to standard vaccine strains, and some laboratories provide comparison to a standard reference panel of wild type PRRSV isolates in a form of dendrogram, or comparison to a production system’s PRRSV sequence database.
Analysis of PRRSV sequences
Sequence similarity, or identity, is determined by aligning two or more sequences using a computer program. An example alignment of several ORF5 sequences from 5 different farms is shown in Figure 1. Pairwise comparisons of their percent identity is shown in the Table. The identities range from 81.2 to 99.8 %. The dendrogram resulting from the phylogenetic analysis shows grouping of the similar sequences (Figure 2). A key question for producers and veterinarians is whether the observed genetic differences between the sequences represent normal variation of a single PRRSV strain in a farm, or represent multiple different strains present in a farm.

Figure 1: Portion of the alignment of ORF5 sequences of PRRSV strains from 5 different farms. From farms 1, 4 and 5, multiple sequences were obtained. Dots in the sequences correspond to positions identical to the PRRSV Type 1 reference strain Lelystad.
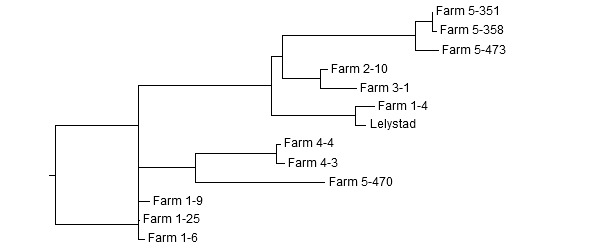
Figure 2: Dendrogram of ORF5 sequences obtained in 5 different farms. Example of interpretation: In Farm 1 two unrelated strains are present. Three sequences are >99% identical to each other, whereas the fourth is only ~83 % related. Instead, it is related to Lelystad virus. The strains from Farm 2 and Farm 3 are closely related (98.2 % identical). Two strains from Farm 4 are closely related (99.5 % identical). In Farm 5 two unrelated strains are present. Three strains are >98 % identical to each other, and about 81 % identical to the fourth one.
Table: Pairwise percent identity between all the aligned ORF5 sequences of a series of type 1 PRRSV samples.
| 1-4 | 1-6 | 1-9 | 1-25 | 2-10 | 3-1 | 4-4 | 4-3 | 5-351 | 5-358 | 5-470 | 5-473 | Lelystad | |
| *** | 83.5 | 83.3 | 83.7 | 93.2 | 92.6 | 86.1 | 86.0 | 88.1 | 88.0 | 83.2 | 88.3 | 98.7 | 1-4 |
| *** | 99.2 | 99.7 | 84.2 | 83.0 | 86.0 | 86.0 | 81.4 | 81.2 | 86.3 | 81.4 | 82.1 | 1-6 | |
| *** | 99.5 | 84.5 | 83.3 | 85.6 | 85.6 | 81.4 | 81.2 | 86.5 | 81.4 | 81.9 | 1-9 | ||
| *** | 84.5 | 83.3 | 86.0 | 86.0 | 81.7 | 81.5 | 86.8 | 81.7 | 82.2 | 1-25 | |||
| *** | 98.2 | 86.6 | 86.5 | 91.1 | 90.9 | 84.2 | 90.4 | 93.3 | 2-10 | ||||
| *** | 84.4 | 84.2 | 90.2 | 90.0 | 83.3 | 89.7 | 92.9 | 3-1 | |||||
| *** | 99.5 | 82.5 | 82.7 | 90.6 | 82.5 | 84.8 | 4-4 | ||||||
| *** | 82.3 | 82.5 | 90.9 | 82.2 | 84.6 | 4-3 | |||||||
| *** | 99.8 | 81.0 | 98.3 | 88.4 | 5-351 | ||||||||
| *** | 81.2 | 98.2 | 88.2 | 5-358 | |||||||||
| *** | 80.9 | 83.2 | 5-470 | ||||||||||
| *** | 88.8 | 5-473 | |||||||||||
| *** | Lelystad |
Interpretation of PRRSV sequences
The sequence of ORF5 has about 600 nucleotides. Different estimates indicate that the overall rate of mutation in this gene is about 0.5 % to 1 % per year.Variation in the rate of genetic change is determined by different non-virus factors. The level of specific and non-specific anti-PRRSV immunity in pigs has a huge impact on viral replication and transmission, exerting a strong inhibitory pressure that diminishes the number of virus copies. Lower viral loads result in reduced transmission rates, further reducing overall viral replication and decreasing the rate of change. The same virus under different host conditions may show different rates of genetic change. Thus, sometimes we can observe higher or lower rates of genetic change, outside of the suggested range of 0.5 % to 1% per year.
The central question in the genetic analysis is whether two sequences are closely related (belong to two variants of the same strain) or are independent (belong to two unrelated strains). It is commonly accepted that PRRSV isolates are related or not, based on a percent similarity cut-off of 97% or 98 %. It is obvious that blind acceptance of a 2% or 3% genetic difference between two isolates, without incorporation of additional knowledge, can lead to erroneous conclusions. The differences between the variants of a single strain circulating in a population for several years will exceed this cut-off eventually. The quality of a relationship interpretation is strengthened by access to additional information, including dates and locations of isolation. It is very important to analyze new PRRSV sequences against a broad reference set representing the farm, the system and the region, as well as representing the global genetic diversity.
The DNA sequencing of PRRSV strains can indicate close relationship or independence of the strains (Figure 2) but cannot be used to predict or explain immunological protection or PRRS outbreaks in immune herds. Also, DNA sequencing does not allow prediction of clinical outcome of infection with a given strain, since genetic markers of virulence have not been identified.
At present, many regional PRRS control or elimination projects are underway, mostly in North America but also in Europe. A comprehensive picture of viral variation at the beginning of control and elimination projects is critical for effective monitoring of progress and effectiveness of implementation procedures, and for identification of new virus introductions to farms and to the region.




")